Entering one or more sequences in a required format using one of two methods:
The sequence submission box.
A file selected using the file upload box:
Optionally selecting:
One or more models in the Model Section box.
One or more options from the Other Options box.
Clicking on the submit button:
NeuroPred will attempt to predict cleavage sites in the submitted sequences and perform additional functions as determined by the different options selected. The output is explained in the Output Documentation.
Optionally, clicking on the button provides a range of enhanced options that control the input and output of NeuroPred. The simplified form can be accessed just by selecting the button. The simplified form is generally the same as the more advanced form with the main defaults automatically selected. Therefore, there is no difference between using the simplified form and the advanced form with the default settings.
Changing the default settings allows NeuroPred to use multiple models, calculate model accuracy statistics and obtain the mass of predicted peptides. The following sections explain the various settings of NeuroPred that can be changed.
A sequence is the only required input to NeuroPred and is the same for either the simplified or advanced option. Sequences that are in the required format may be entered into the sequence submission box, or uploaded from a text file; multiple sequences can be submitted at the same time. An uploaded sequence file takes priority over those entered in the sequence submission box; therefore, only sequences in the file will be used, even if sequences were also entered into the sequence submission box.
There are two supported formats that are based on the FASTA format: the Basic Format and the Known Cleavage Format, which is an extension of the Basic Format that includes known cleavage information.
Basic Format: This format begins with a single-line description or header with a greater-than (">") symbol in the first column that is followed by lines of sequence data. The sequence can be provided in either upper or lower case on a new line following the header line. Spaces or other white space characters (such as tabs or new lines) can be included such that the sequence may span several lines but other non-sequence characters will result in an incorrect sequence. An example of the Basic Format for the Human proglucagon precursor sequence is:
Known Cleavage Format: An extension of the Basic Format that includes the known cleavage sites. This format is required in order to compute model accuracy statistics. The known cleavage sites must be entered immediately after the sequence on a new line. For every amino acid in the sequence a corresponding zero (0) or one (1) must be present where 0 and 1 denote non-cleavage and cleavage, respectively. All cleavages are assumed to occur C-terminal to the indicated amino acid. This binary sequence must be the same length as the sequence and, like the sequence, can include white space characters (such as spaces, tabs and new lines) and can span multiple lines.
Sequence Entry Errors:NeuroPred will automatically ignore any sequences in incorrect formats and the error message, "There were either no sequences entered or sequences were entered in the wrong format" is displayed when there are no valid sequences submitted.
Known Cleavage Format: A warning message, "Warning: Cannot do model accuracy because either there is no data or something is wrong with the data format" is displayed if at least one sequence has an incorrect format. In this situation, NeuroPred will only predict cleavage sites and display the associated output. An additional warning message is displayed when there is a mismatch between the sequence and the binary input.
Other possible sequence input errors that may cause unexpected results include sequences that do not start on a new line immediately after the header, the header line spanning more than one line, or non-standard characters.
The model selection options are the same in both interfaces; however, the advanced options interface provides a wider range of models. All models except the Known Motif were trained on specific organisms or related species. Generally the most correct predictions are obtained when the model is trained either on the same or related species. However, because no single prediction model is ideal, the simultaneous use of complementary methods is allowed and encouraged. The Model Accuracy Statistics from the Output Selection Task can be used to assess the performance of different models to assist in selecting the appropriate models.
The following four cleavage prediction models are available in both interfaces, the first model is an empirical model, whereas all other models are binary logistic regression models.
The following models are available for both interfaces:
Known Motif:
This model, reported by Southey et al. (2006a), is based on the occurrence of the following motifs in the sequence: xxKR, xxKK, xxRR, RxxR and RxxK, where x is any amino acid and cleavage occurs after the right-most or C-terminal amino acid. When any of these motifs is present, the cleavage probability is set to 0.88 and when two of these motifs are present (e.g. RxKR, which is composed of the motifs xxKR and RxxR), the cleavage
probability is set to 0.997. This is the default model.
Mollusc:
The Complex Model, reported by Hummon et al. (2003), trained on precursor sequences from the mollusc, Aplysia californica.
Mammalian:
Model reported by Amare et al. (2006), trained on published mammalian precursor sequences from cow, human, mouse, pig and rat.
Insect:
A joint model derived from the models trained by Southey et al. (2008) on published precursor sequences from the honey bee, Apis mellifera and fruit fly, Drosophila melanogaster.
The following binary logistic regression models are only available with the advanced option interface:
Mollusc_Basic:
Model reported by Hummon et al. (2003), trained on precursor sequences from the mollusc, Aplysia californica. This model has fewer terms than the Mollusc model and also had poorer predictive ability than the Mollusc mode (Hummon et al. 2003).
Apis:
Model reported by Southey et al. (2008), trained on published precursor sequences from the honey bee, Apis mellifera, reported by Hummon et al., 2006.
Drosophila:
Model reported by Southey et al. (2008), trained on precursor sequences from the fruit fly, Drosophila melanogaster
A model trained on published rat, Rattus norvegicus, precursor sequences by Tegge et al. (2007, 2008).
Any Basic Site:
Simply considers any basic amino acid (Arginine or Lysine) as cleaved. However, when the amino acid combinations listed in the known motif model (e.g. xxKR, xxKK, xxRR, RxxR and RxxK) are present, only the last amino acid in the motif is considered as cleaved.
The newer version of NeuroPred, Test 2009
permits the use of following models under the advanced option interface that include amino acid physicochemical properties and models trained
using artifical neural networks. The insect models were developed by Southey et al. (2008) and the
human, mouse, bovine and rat models were developed by Tegge et al. (2007, 2008) :
Bovine amino acids plus properties Log Reg:
Logistic regression model trained on bovine sequences that uses both amino acids and amino acid physicochemical properties.
Human amino acids plus properties Log Reg:
Logistic regression model trained on human sequences that uses both amino acids and amino acid physicochemical properties.
Mouse amino acids plus properties Log Reg:
Logistic regression model trained on mouse sequences that uses both amino acids and amino acid physicochemical properties.
Rat amino acids plus properties Log Reg:
Logistic regression model trained on rat sequences that uses both amino acids and amino acid physicochemical properties.
Apis Neural Net:
Artifical neural network model trained on Apis sequences that uses only amino acids.
Drosophila Neural Net:
Artifical neural network model trained on Drosophila sequences that uses only amino acids.
Bovine amino acids only Neural Net:
Artifical neural network model trained on bovine sequences that uses only amino acids.
Human amino acids only Neural Net:
Artifical neural network model trained on human sequences that uses only amino acids.
Mouse amino acids only Neural Net:
Artifical neural network trained on mouse sequences that uses only amino acids.
Rat amino acids only Neural Net:
Artifical neural network model trained on rat sequences that uses only amino acids.
Bovine amino acids plus properties Neural Net:
Artifical neural network model trained on bovine sequences that uses both amino acids and amino acid physicochemical properties.
Human amino acids plus properties Neural Net:
Artifical neural network model trained on human sequences that uses both amino acids and amino acid physicochemical properties.
Mouse amino acids plus properties Neural Net:
Artifical neural network trained on mouse sequences that uses both amino acids and amino acid physicochemical properties.
Rat amino acids plus properties Neural Net:
Artifical neural network model trained on rat sequences that uses both amino acids and amino acid physicochemical properties.
The binary logistic regression models vary in both the number of locations surrounding the cleavage site and the specific amino acids at these locations. Details on the specific composition of the models as well as description of training data and performance of each model can be found in the respective references. The general differences between the models are summarized in the following table.
Summary of Prediction Models
Model
Window Length
Location of Cleavage Site
Number of Amino Acid-Location Combinations Used
Example of Window
Known Motif
4
4
4
DSRR
Mollusc
13
5
10
LDSRRAQDFVQWLM
Mammalian
18
9
11
YSKYLDSRRAQDFVQWLM
Insect
15
6
20
YLDSRRAQDFVQWLM
Mollusc_Basic
13
5
24
LDSRRAQDFVQWLM
Apis
7
4
9
DSRRAQD
Drosophila
15
6
20
YLDSRRAQDFVQWLM
Bovine
18
9
67
YSKYLDSRRAQDFVQWLM
Human
18
9
34
YSKYLDSRRAQDFVQWLM
Mouse
18
9
101
YSKYLDSRRAQDFVQWLM
Rat
18
9
71
YSKYLDSRRAQDFVQWLM
Description of Summary of Prediction Models Columns:
Window Length:
Number of locations surrounding the site used by the model to predict cleavage. This is typically smaller
than the number of locations used in training the model.
Location of Cleavage Site:
The location of the cleaved
site in the window from the left-hand or N-terminal end of the window.
Number of Amino
Acid-Locations Combinations Used:
This is the maximum number of
possible combinations of amino acids and locations within the window that
are used by the model. A model can have many amino acids at the same
location.
Example of Window:
An example of a possible window
with the cleavage site denoted by the bold amino acid for the same sample
sequence such that actual cleavage occurs to the right or C-terminal to
this amino acid.
Predicts cleavage sites for submitted
sequences using the selected cleavage prediction models. This is the default task.
Obtain Mass of Predicted Peptides:
Predicts cleavage sites for submitted
sequences using the selected cleavage prediction models and calculates
the average and monoisotopic molecular
masses of predicted peptides resulting from cleavage and any selected
post-translational modifications.
Model Accuracy Statistics:
Predicts cleavage sites for submitted sequences
and calculates model accuracy statistics for all submitted sequences
and selected cleavage prediction models. In order to obtain model
accuracy statistics, the known cleavage format is required. A warning
message, "Warning: Cannot do model accuracy because either
there is no data or something is wrong with the data format" is provided if at least one sequence has an incorrect format and model
accuracy statistics are not computed for any sequence. See Output
Documentation for further details.
Print Probabilities of Basic Sites Only:
Only prints the predicted
probabilities of the basic sites within the submitted sequences.
A variety of options are available to control the input and output of NeuroPred depending on whether the
simplified or advanced option interface is used.
1. Simplified Options Interface
Other Options
Display Cleavage Probabilities?
(select for yes)
Input the length of the signal peptide (use zero (0) for no signal peptide)
Sort the output from mass calculations on
Remove any C-terminal K and R from predicted peptides?
(select for yes)
Select Post-Translational Modifications (PTMs)
Description of Other Options:
Display Cleavage Probabilities?
This option permits the display
of the actual probabilities calculated for the basic amino acids in the
sequence. This option is unselected by default so the probabilities are not displayed unless this box is selected.
Input the length of the signal peptide:
This option provides the
default length of the signal peptide when a Signal length identifier is not used. A default value of 15 is used as determined by the smallest
known example in a wide range of known precursors. Use zero when there is
no signal peptide present such as for sequence fragments.
Sort the output from mass calculations on:
This option is only valid when
the Obtain Mass of Predicted Peptides task is selected and,
therefore, it is ignored with all other tasks. By default the predicted
peptides and associated masses are not sorted but this option permits the
output to be sorted by different criterion. The predicted peptides can be
sorted from low to high by Average mass or Monoisotopic mass, or by the location of the predicted peptide Sequence based
on location in the original input sequence and any post-translational
modifications applied to that peptide.
Remove any C-terminal K and R from predicted
peptides?
This option is only valid when
the Obtain Mass of Predicted Peptides task is selected and,
therefore, it is ignored with all other tasks. This option permits the
removal of C-terminal basic amino acids (Arginine and Lysine) from the cleaved sequence and is selected by default.
Select Post-Translational Modifications (PTMs):
This option is only valid when
the Obtain Mass of Predicted Peptides task is selected and,
therefore, it is ignored with all other tasks. This option permits the
selection of different groups of post-translational modifications (See Section
F. Selection of Post-Translational Modifications for more details):
No PTMs selected:
No post-translational modifications are selected and is
the default selection.
Common PTMs:
Common post-translational modifications (Amidation, Pyroglutamylation, Acetylation and Tyr-Sulfation) are applied.
All PTMs:
All post-translational modifications listed under the Advanced Options Interface are applied.
Number of amino acids surrounding cleavage site: Before
After
Number of amino acids in the signal sequence:
Signal length identifier:
Lower mass value (Da):
Upper mass value (Da):
Maximum number of amino acids in a peptide:
Degree of peptide extension:
Sort the output from mass calculations on
Ignore processing rules:
Use basic sites for accuracy statistics?
Description of Modeling and Mass Calculation Options:
Threshold cleavage probability:
The minimum value that the
predicted cleavage probability must exceed to declare the associated site
as being cleaved. The default threshold cleavage probability is 0.50 and
can range from 0 to 1.0. A low threshold cleavage probability will
increase the number of true positives, while a higher threshold cleavage
probability will increase the number of true negatives.
Confidence interval significance level:
The significance level, α,
required to generate a 100*(1-α)% confidence interval. The default α value is 0.05 and it can
range from 0.005 to 0.2. The α value
of 0.05 means that the true cleavage probability lies within the confidence
interval range in 95% of the samples. The confidence interval range can be
used as an indicator of the uncertainty associated with cleavage prediction
where a narrow interval range implies reliable prediction and a wider range
implies a less reliable prediction.
Number of amino acids surrounding cleavage site:
Specifies the minimum number of
amino acids that must surround the cleavage site for cleavage to occur and
is currently used to avoid predicting cleavage near the sequence termini.
The Before option denotes the number of amino acids
N-terminal to the cleavage site and the After option denotes the number of amino acids C-terminal to the cleavage site.
The default values are based on empirical observation of known cleavages
and information from the crystal structure of furin.
Signal peptide length:
Located at the N-terminus of a
precursor, the signal peptide is a transport marker needed for the
translocation of the precursor into the lumen of the endoplasmic reticulum
and is co-translationally removed by a signal
peptidase. Consequently, this region must be removed prior to prediction of
cleavage sites. The predicted length of the signal peptide for any sequence
can be obtained using the SignalP application. There are two ways to account
for the signal peptide:
Number of amino acids in
the signal sequence:
This option specifies a global
length of the signal peptide for all submitted sequences so that no
prediction occurs in this region. The default value is 15 but can range
from 0 to the length of the sequence. If an invalid value is found, NeuroPred defaults to signal peptide
length of 15.
Signal length
identifier:
An individual length for each
sequence can be indicated in the header of the FASTA format by the
"Signal length identifier". The default signal length identifier
is SignalP= and can be changed to
any value. The presence of a valid value using this identifier will
override the default global length of the signal peptide; thus, this
specification is useful when there are multiple sequences that have varying
signal peptide lengths. The signal length identifier is case insensitive
such that SIGnalP is treated the same as signalp. The number of amino acids in the signal
sequence must immediately follow this identifier and a space must occur
after the number of amino acids. For example, "SignalP=25 aa" is valid specification but "SignalP=25aa" is invalid. If an invalid value
is found, NeuroPred uses the default value
of 15.
Lower mass value:
Predicted peptides with masses lower than this value are not reported. The default value is 0 Da.
Upper mass value:
Predicted peptides with masses higher than this value are not reported. The default value is 15,000 Da.
Maximum number of amino acids in a peptide:
Does not output cleaved
peptides that have more amino acids than this value, thus limiting the
number of reported final products. The default is set to 1000 amino acids.
Degree of peptide
extension:
In order to account for false
positive (cleaved) predictions, the predicted peptide can be extended to
include as many adjacent peptides as specified in the "Degree of
peptide extension" box. This option greatly increases the number
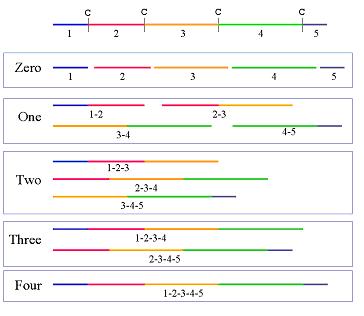
of peptides in the output but can be useful for peptidomics studies.The figure below
illustrates how extending the peptide increases the combinations of
predicted peptides. The degree of peptide extension can range from 0 to any
integer. As shown in the example, a zero degree of extension produces the
shortest possible peptides whereas higher degrees of extension produce
longer peptides. When the degree of extension exceeds or is equal to the
total number of predicted cleavage sites, all possible peptides except the
original sequence are produced. Consequently, in the example shown below
for an extension value of three or higher, all extensions up to but not
including the fourth extension are provided in the output.
Peptide
extension increases the number of predicted peptides
Sort the output from mass calculations on:
This option is only valid when
the Obtain Mass of Predicted peptides task is selected and,
therefore, it is ignored with all other tasks. By default the predicted
peptides and associated masses are not sorted but this option permits the
output to be sorted by different criterion. The predicted peptides can be
sorted from low to high Average mass or Monoisotopic mass, or by the location of the predicted peptide Sequence based
on location in the original input sequence and any post-translational
modifications applied to that peptide.
Ignore processing rules:
This option determines whether or not the processing rules described by Amare et al. 2006 and Southey et al. 2008 are implemented. By default these rules are followed such that cleavage sites are ignored when cleavage could occur at a nearby location (specifically where a basic amino acid was present in the P1 location, a basic amino acid was present in either the P1' or P4' locations, and there was no basic amino acid located in either the P2 or P4 locations, where "P" and "P'" denotes N-terminal and C-Terminal locations relative tot he cleavage site, respectively, such that cleavage occurs between P1 and P1' locations). The ignored sites are termed redundant sites and are denoted as 'r' in the Cleavage Prediction diagram in the output of NeuroPred.
Use basic sites for accuracy statistics?:
This option is only valid when
the Model Accuracy Statistics task is selected and, therefore, it is ignored with all other tasks. By default the model accuracy statistics are calculated only with basic amino acids. Selecting No to this option permits the model accuracy to be calculated over the complete sequence including all non-basic sites that are usually considered uncleaved.
Prohormone precursors undergo extensive modification before active
neuropeptides and hormones are obtained. In addition to cleavages at basic
sites and immediate removal of C-terminal basic residues (Trim
C-terminal K and R), several other modifications may be present. The
most common PTMs are amidation (of C-terminal glycine) and pyroglutamylation (cyclization of N-terminal glutamate or glutamine). Sulfation of tyrosine (Tyr-Sulfation) and acetylation are also common, albeit occurring somewhat less frequently; thus these four
common PTMs are grouped together while even less
common PTMs are grouped separately. Disulfide
bond formation between two cysteine residues
resulting in a mass loss of 2 Da is a common PTM
in neuropeptides, but it is difficult to predict whether the disulfide bond
is formed between two peptides or within a peptide containing two or more cysteines. In addition, it is difficult to identify the cysteine pairs involved in bond formation. For
these reasons, disulfide bond formation is not modeled in NeuroPred. The user may consult
web-based tools such as Cyspred to determine potentially disulfide bonding cysteines. A table of the available PTMs can be found here.
The advanced option interface permits the selection (or deselection)
of individual PTMs where Amidation and Pyroglutamylation are selected by default.
Amare, A., Hummon, A.B., Southey, B.R., Zimmerman, T.A., Rodriguez-Zas, S.L., Sweedler, J.V., Bridging neuropeptidomics and genomics with bioinformatics: prediction of mammalian neuropeptide prohormone processing. J. Proteome Res. 2006, 5, 1162-1167. Abstract.
Hummon, A.B., Hummon, N.P., Corbin, R.W., Li, L.J., Vilim, F.S., Weiss, K.R., Sweedler, J.V., From precursor to final peptides: a statistical sequence-based approach to predicting prohormone processing. J. Proteome Res. 2003, 2, 650-656. Abstract.
Hummon, A.B. Richmond, T.A. Verleyen, P. Baggerman, G. Huybrechts, J. Ewing, M A. Vierstraete, E. Rodriguez-Zas, S.L. Schoofs, L. Robinson, G.E. Sweedler, J.V. , From the Genome to the Proteome: Uncovering Peptides in the Apis Brain, Science2006, 314, 647-649. Abstract.
Southey, B.R., Rodriguez-Zas, S.L., Sweedler, J.V., Prediction of neuropeptide prohormone cleavages with application to RFamides. Peptides 2006a, 27, 1087-1098. Abstract.
Southey B.R., Amare A., Zimmerman T.A., Rodriguez-Zas S.L., Sweedler J.V., NeuroPred: a tool to predict cleavage sites in neuropeptide precursors and provide the masses of the resulting peptides. Nucleic Acids Res. 2006b, 34 (Web Server issue), W267-272. Abstract.
Tegge, A.N. Southey, B.R. Sweedler, J.V. Rodriguez-Zas, S.L., Enhanced Prediction of Cleavage in Bovine Precursor Sequences. Lecture Notes in Computer Science, Bioinformatics Research and Applications, Vol. 4463, pp. 350-360, 2007, Springer. Abstract.
Southey, B.R., Hummon, A.B., Richmond, T.A., Sweedler, J.V., Rodriguez-Zas, S.L., Prediction of neuropeptide cleavage sites in insects. Bioinformatics, 2008, 24, 815-825. Full Text